 Douleur - Allodynie/Hyperalgésie Thermique
Douleur - Allodynie/Hyperalgésie Thermique Douleur - Spontanée - Déficit de Posture
Douleur - Spontanée - Déficit de Posture Douleur - Allodynie/Hyperalgésie Mécanique
Douleur - Allodynie/Hyperalgésie Mécanique Apprentissage/Mémoire - Attention - Addiction
Apprentissage/Mémoire - Attention - Addiction Physiologie & Recherche Respiratoire
Physiologie & Recherche Respiratoire





























 Douleur
Douleur Système Nerveux Central (SNC)
Système Nerveux Central (SNC)  Neurodégénérescence
Neurodégénérescence Système sensoriel
Système sensoriel Système moteur
Système moteur Troubles de l'humeur
Troubles de l'humeur Autres pathologies
Autres pathologies Système musculaire
Système musculaire Articulations
Articulations Métabolisme
Métabolisme Thématiques transversales
Thématiques transversales Congrès & Meetings
Congrès & Meetings Authors
A Kubota, M Juanola-Falgarona, V Emmanuele et al
Lab
Department of Neurology, Columbia University Medical Center, New York, USA
Journal
Human Molecular Genetics
Abstract
X-linked scapuloperoneal myopathy (X-SM), one of Four-and-a-half LIM 1 (FHL1) related diseases, is an adult-onset slowly progressive myopathy, often associated with cardiomyopathy. We previously generated a knock-in mouse model that has the same mutation (c.365 G > C, p.W122S) as human X-SM patients. The mutant male mouse developed late-onset slowly progressive myopathy without cardiomyopathy. In this study, we observed that heterozygous (Het) and homozygous (Homo) female mice did not show alterations of skeletal muscle function or histology. In contrast, 20-month-old mutant female mice showed signs of cardiomyopathy on echocardiograms with increased systolic diameter [wild-type (WT): 2.74 ± 0.22 mm, mean ± standard deviation (SD); Het: 3.13 ± 0.11 mm, P < 0.01; Homo: 3.08 ± 0.37 mm, P < 0.05) and lower fractional shortening (WT: 31.1 ± 4.4%, mean ± SD; Het: 22.7 ± 2.5%, P < 0.01; Homo: 22.4 ± 6.9%, P < 0.01]. Histological analysis of cardiac muscle revealed frequent extraordinarily large rectangular nuclei in mutant female mice that were also observed in human cardiac muscle from X-SM patients. Western blot demonstrated decreased Fhl1 protein levels in cardiac muscle, but not in skeletal muscle, of Homo mutant female mice. Proteomic analysis of cardiac muscle from 20-month-old Homo mutant female mice indicated abnormalities of the integrin signaling pathway (ISP) in association with cardiac dysfunction. The ISP dysregulation was further supported by altered levels of a subunit of the ISP downstream effectors Arpc1a in Fhl1 mutant mice and ARPC1A in X-SM patient muscles. This study reveals the first mouse model of FHL1-related cardiomyopathy and implicates ISP dysregulation in the pathogenesis of FHL1 myopathy.
BIOSEB Instruments Used
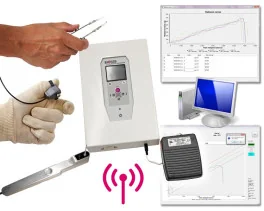
Grip strength test (BIO-GS3)
Source :
